新手总是查不到详细一点的蛋白纯化资料,苦不堪言。课本上内容一带而过,文献里一笔了事,一遇到蛋白纯化总是混乱不堪。这节介绍一下4大常用蛋白纯化技术的原理、实操步骤以及注意要点,来吧,一起学习学习。
一、盐析法-硫酸铵沉淀
1.原理
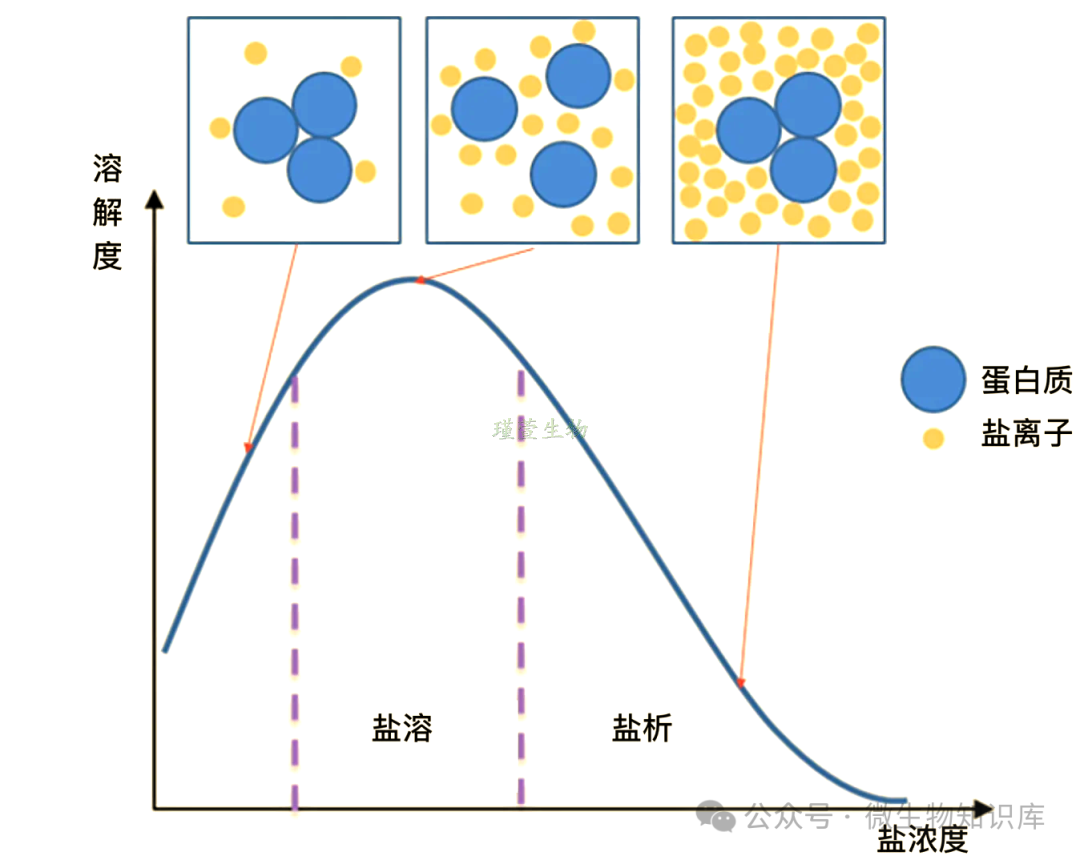
利用不同蛋白质在不同浓度中性盐溶液中的溶解度差异进行分离。向蛋白质溶液中加入中性盐(如硫酸铵),盐离子与水分子结合,破坏蛋白质的水化膜,同时抑制蛋白质弱电解质的解离,使蛋白质带电荷减少,更容易聚集析出,从而实现蛋白质的沉淀和分离。
不同蛋白沉淀盐浓度不同,可实现分级分离、初步除杂。
2.试剂
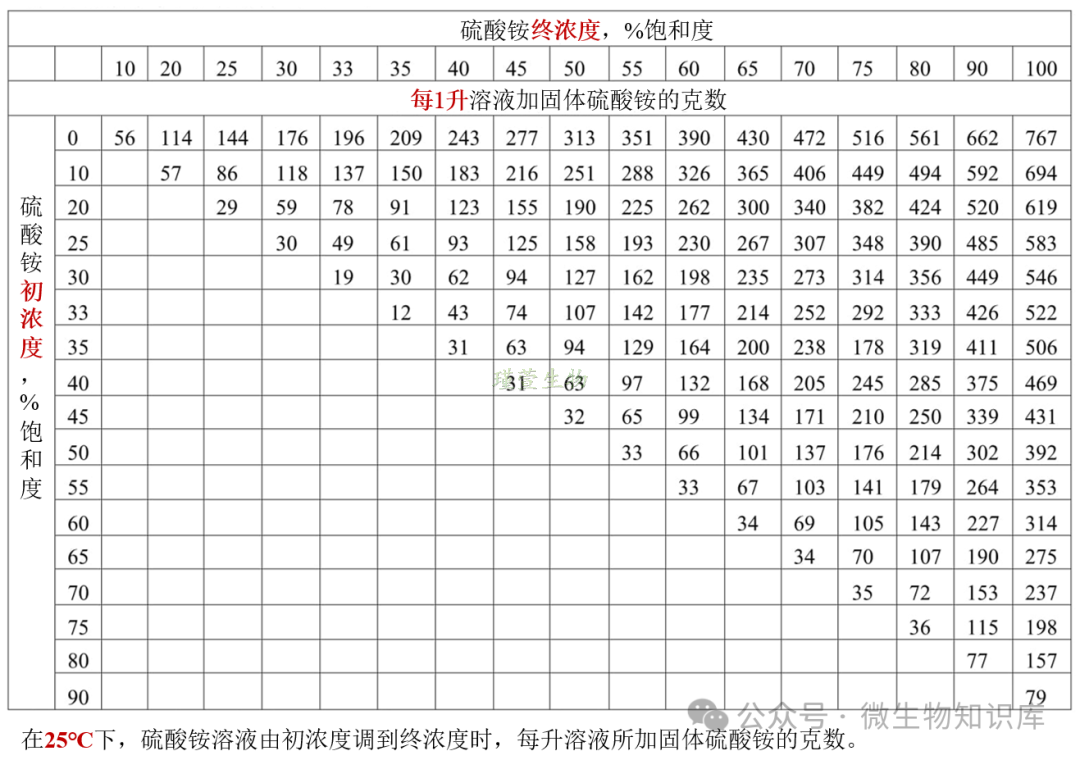
主要计算硫酸铵的用量。
硫酸铵饱和度:在某温度下,溶液中硫酸铵的溶解量占该温度下最大可溶解量的百分比。
比如,温度25°C,目标饱和度为50%,100m蛋白溶液,查表可得25°C下50%=313g/L,100mL溶液需要加:313g/LX0.1L=31.3g。
3.操作步骤
(1)准备样品:低温破碎裂解细胞后,离心取上清;
(2)准备硫酸铵:准备硫酸铵粉末,初步计算硫酸铵用量;
(3)添加硫酸铵:磁力搅拌条件下,缓慢加入硫酸铵;
(4)沉淀蛋白:连续搅拌30min,溶液中会出现浑浊,沉淀蛋白;
(5)收集蛋白:4℃,13000rpm离心,去上清,收集沉淀,沉淀即为蛋白;
(6)洗涤沉淀:用缓冲液或去离子水洗涤沉淀,离心后去上清,收集沉淀;
(7)溶解沉淀:用适合的缓冲液溶解蛋白沉淀,使蛋白完全溶解;
(8)透析脱盐:利用透析袋去除残留硫酸铵。
(9)检测:利用SDS-PAGE蛋白电泳检测纯化蛋白。
4.注意事项⚠️
(1)蛋白质易变性,操作过程既要注意温度,也要注意硫酸铵在不同温度时的溶解度。
(2)硫酸铵必须少量多次添加,边搅边加,添加太快会导致局部硫酸铵浓度过大,析出不同的蛋白,无法分离蛋白。
(3)盐析后必须透析除盐,否则影响后续挂柱。
(4)该方法只是粗提,无法分离溶解度相近的蛋白。
(5)如果不清楚硫酸铵沉淀蛋白浓度,可以向上清中继续加入硫酸铵粉末,继续沉淀蛋白,连续沉淀几个梯度,利用SDS-PAGE蛋白电泳检测,选择合适的浓度沉淀相应蛋白。
二、亲和层析-镍柱亲和层析
1.原理
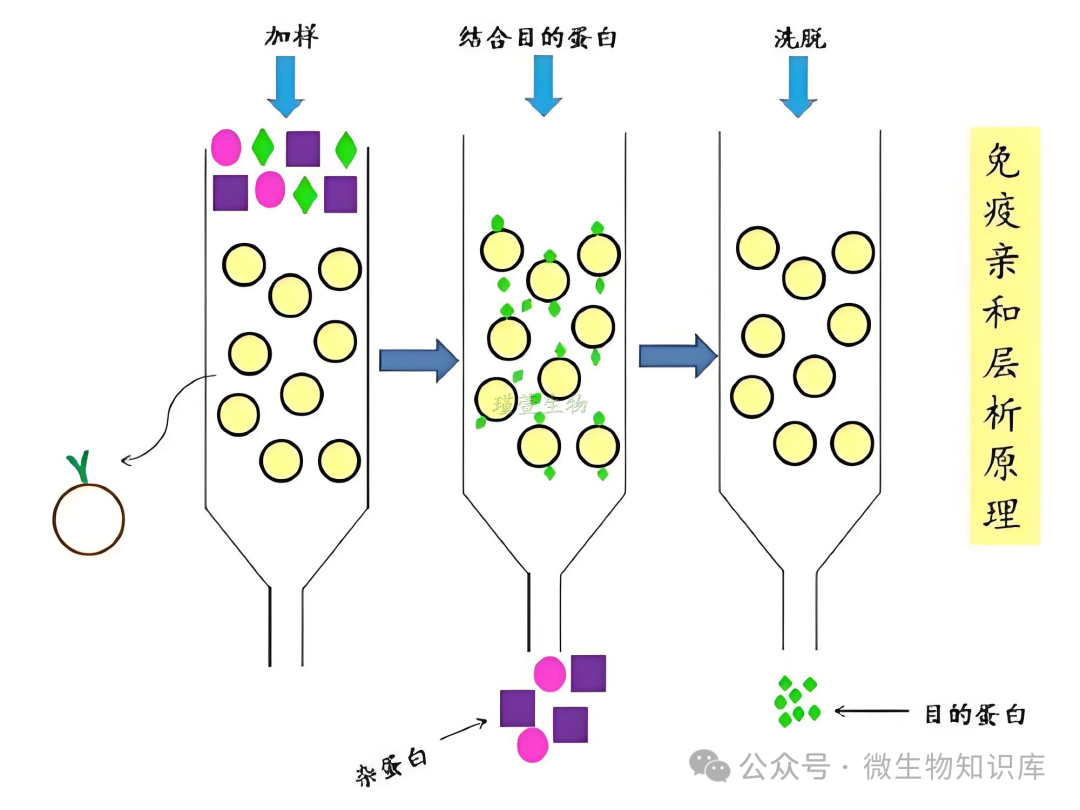
利用化学结合的特异性,将目标分子(如His-tag)与固定在层析介质上的镍离子(Ni2+)之间的特异性结合,通过亲和层析轻松地从细胞裂解液中分离出目标蛋白。
因His标签蛋白中的组氨酸残基侧链含有咪唑基团,可与Ni2+形成配位键,实现特异性吸附。通过调节缓冲液中咪唑浓度竞争性洗脱目标蛋白,低浓度咪唑洗去杂蛋白,高浓度咪唑破坏His-tag与Ni2+的结合,释放目标蛋白。
因此,该方法可实现目标分子的分离纯化,特异性强、纯化倍数高。
2.试剂
(1)裂解缓冲液:20mM Tris-HCI (pH8.0),500mM NaCI,5mM咪唑,1mMPMSF(蛋白酶抑制剂),1mg/mL溶菌酶(可选);
(2)平衡缓冲液:20mM Tris-HCI (pH8.0),500mM NaCI,5mM咪唑。
(3)洗杂缓冲液:20mM Tris-HCI (pH8.0),500mM NaCI,20-50 mM咪唑(梯度优化)。
(4)洗脱缓冲液:20mM Tris-HCI (pH 8.0),500mM NaCI,250-500mM 咪唑。
(5)Ni-NTA亲和层析柱
3.操作步骤
(1)准备样品:低温破碎裂解细胞后,离心取上清;注:跟上面不同的是,这里的蛋白必须有His标签。
(2)镍柱预处理:将Ni-NTA填料装柱(或使用预装柱),用5倍柱体积(CV)去离子水冲洗。用5CV平衡缓冲液平衡柱子至基线稳定(pH和电导率与样品一致);
(3)上样与结合:将样品以0.5-1mL/min流速缓慢上样至柱,确保His标签蛋白充分结合,收集流穿液(用于后续分析是否结合完全);
(4)洗杂蛋白:用5-10CV平衡缓冲液冲洗未结合的杂蛋白。逐步增加咪唑浓度(如20mM一50mM)洗去弱结合杂蛋白,监测A280至基线平稳;
(5)洗脱目标蛋白:使用含250-500mM咪唑的洗脱缓冲液洗脱目标蛋白,收集洗脱峰(A280显著升高时),分多管收集,标记为Elution1、Elution2等;
(6)柱再生与保存:用5V含500mM咪唑的缓冲液清洗柱子,去除残留蛋白。用5V去离子水冲洗后,以20%乙醇保存镍柱(4°C);
(7)检测:利用SDS-PAGE蛋白电泳检测纯化效果。
4.注意事项⚠️
(1)蛋白样品必须澄清,严禁浑浊上样,容易堵柱。
(2)体系避免高浓度EDTA、强还原剂,会脱镍、挂柱失败。
(3)上样流速不能过快,否则挂柱不完全。
(4)梯度洗杂+高浓度洗脱,不要一步高浓度洗脱。
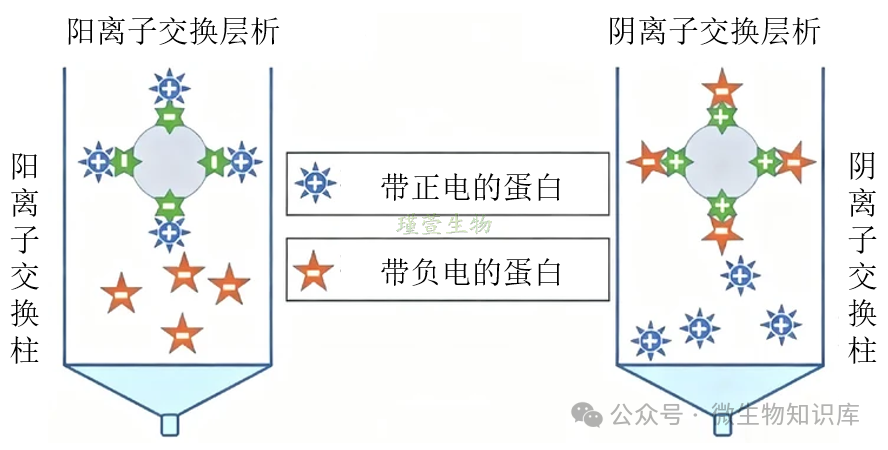
三、离子交换层析
1.原理
根据蛋白分子的表面电荷特性分离和纯化目标蛋白的方法,通过蛋白与带相反电荷的固定相(离子交换介质)之间的静电相互作用实现分离。
蛋白在不同pH条件下的表面电荷由其等电点(pI)决定:pH<pl,蛋白带正电;pH>pl,蛋白带负电。
阳离子交换层析:介质带负电,吸附正电荷蛋白。
阴离子交换层析:介质带正电,吸附负电荷蛋白。
通过连续改变洗脱环境的pH或盐浓度,创造竞争条件,使结合力不同的蛋白质依次被替换下来,达到分离目的。
2.试剂
(1)平衡液A:低盐结合缓冲液(20~50 mM Tris/PB)
3.操作步骤
(1)准备样品:低温破碎裂解细胞后,离心取上清;
(2)选择层析柱:根据蛋白等电点选择对应离子柱;
(3)装柱:干粉树脂预处理,装入层析柱,自然沉降,柱床平整;
(4)柱平衡:先走纯水3-5 倍柱体积,再用平衡液 A冲洗5-10倍柱体积,流出液 pH、电导与平衡液一致即平衡完成;
(5)上样:以流速1-2mL/min,将蛋白样品上样到离子交换柱上,
(7)梯度洗脱:用不同pH值或离子强度的缓冲液进行梯度洗脱,收集洗脱液,检测其中的蛋白质;
(8)柱再生与保存:用0.5M NaOH 浸泡或冲洗 3-5 倍柱体积,用纯水冲至中性,20% 乙醇封存,4℃保存。
4.注意事项⚠️
(1)上样样品必须低盐,高盐直接无法挂柱。
(2)缓冲液pH必须稳定,波动会导致吸附异常。
(3)禁止在pI附近操作,蛋白极易沉淀聚集。
(4)优先线性梯度洗脱,分离效果远优于一步洗脱。
(5)若目标蛋白直接流穿,说明pH选错 ,重新配制。
(6)该方法分辨率较高,可通过调整缓冲液的pH值和离子强度来优化分离效果,适用于各种带电蛋白质的分离,且可处理较大体积的样品。
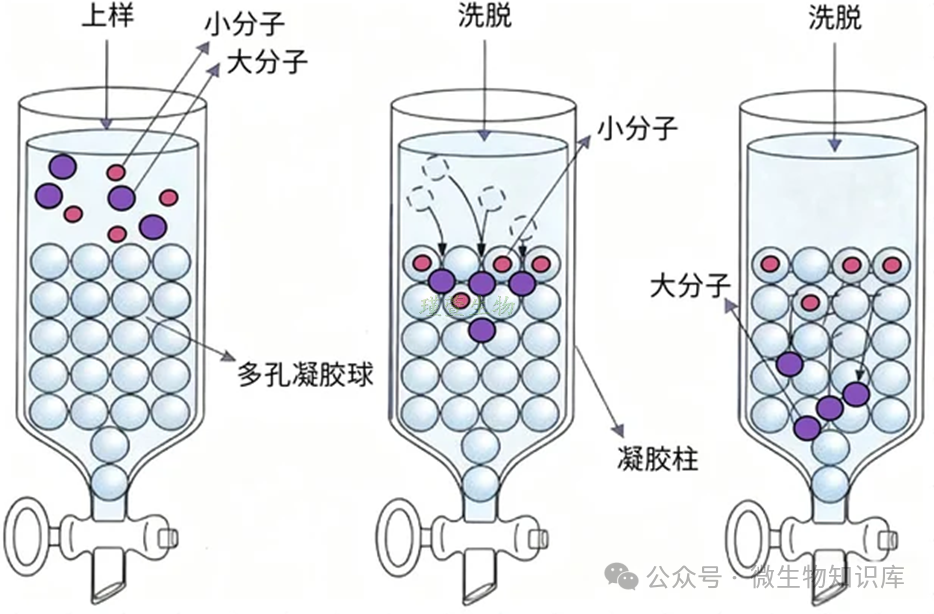
四、凝胶过滤层析-分子筛
1.原理
2.试剂
(1)缓冲液:PBS、Tris-HC:
(2)选凝胶型号:
Sephadex G-75:分离3k-70k Da蛋白
(1)准备样品:低温破碎裂解细胞后,离心取上清;
(2)选择凝胶型号:根据蛋白大小选择;
(3)装柱:干粉树脂预处理,装入层析柱,自然沉降,柱床平整;
(4)柱平衡:洗脱缓冲液匀速冲洗3~5 倍柱体积,流出液 pH、电导与平衡液一致即平衡完成;
(5)上样:缓慢将蛋白样品上样到凝胶柱,待样品全部渗入胶床后,立刻加少量缓冲液封住液面;
(8)柱再生与保存:洗完直接用 3~5 倍柱体积缓冲液冲净即可。
4.注意事项⚠️
(1)上样体积不能过大,否则分辨率暴跌。
(2)流速必须缓慢平稳,速过快分子来不及筛分,分离不纯。
(3)有聚集体一定要提前去除,聚合体最先出峰。
(4)不适合分离分子量接近的蛋白。
(5)该方法只按分子量大小分离,与蛋白电荷、极性无关。
(6)柱子必须垂直放置,柱床压实均匀,不能出现气泡、断层、裂纹。
五、总结
1.盐析法粗提。
2.亲和层析或离子交换层析去除大部分杂蛋白。
3.凝胶过滤层析进行精细纯化。
Copyright © 2022-2030 武汉瑾萱生物科技有限公司 版权所有 地址:武汉东湖新技术开发区光谷三路777号自贸生物创新港B区(生物医药平台检验研发楼) 备案号:鄂ICP备2022008796号 网站地图